


有疑问、问专家
提问
 E邀专家
E邀专家

》等五项技术指导原则公开征求意见的通知")
为持续完善仿制药个药指导原则,进一步规范乙酰半胱氨酸颗粒、依托咪酯乳状注射液、依帕司他片、硫辛酸片和利丙双卡因乳膏的生物等效性研究,经广泛调研和讨论,我中心组织起草了《乙酰半胱氨酸颗粒生物等效性研究指导原则(征求意见稿)》等五项技术指导原则。
我们诚挚地欢迎社会各界对征求意见稿提出宝贵意见和建议,以便后续完善。征求意见时限为自发布之日起1个月。
请将您的反馈意见发到以下联系人的邮箱。
联系人:周誉;安娜
联系方式:zhouy@cde.org.cn;ann@cde.org.cn
感谢您的参与和大力支持。
国家药品监督管理局药品审评中心
2023年7月27日
附件:
1、《乙酰半胱氨酸颗粒生物等效性研究指导原则(征求意见稿)》
2、《乙酰半胱氨酸颗粒生物等效性研究指导原则(征求意见稿)》起草说明
3、《依托咪酯乳状注射液生物等效性研究指导原则(征求意见稿)》
4、《依托咪酯乳状注射液生物等效性研究指导原则(征求意见稿)》起草说明
5、《依帕司他片生物等效性研究指导原则(征求意见稿)》
6、《依帕司他片生物等效性研究指导原则(征求意见稿)》起草说明
7、《硫辛酸片生物等效性研究技术指导原则(征求意见稿)》
8、《硫辛酸片生物等效性研究技术指导原则(征求意见)》起草说明
9、《利丙双卡因乳膏生物等效性研究指导原则(征求意见稿)》
10、《利丙双卡因乳膏生物等效性研究指导原则(征求意见稿)》起草说明
11、《___________生物等效性研究技术指导原则(征求意见稿)》征求意见反馈表
乙酰半胱氨酸颗粒生物等效性研究技术指导原则
(征求意见稿)
一、概述
乙酰半胱氨酸颗粒(Acetylcysteine Granules)为祛痰药,其化学结构中的巯基可使粘蛋白的双硫键断裂,降低痰粘度,使痰容易咳出。临床上用于慢性支气管炎等咳嗽有粘痰而不易咳出的患者。乙酰半胱氨酸为内源性物质,但体内浓度较低。
乙酰半胱氨酸颗粒生物等效性研究应符合本指导原则要求,还应参照《以药动学参数为终点评价指标的化学药物仿制药人体生物等效性研究技术指导原则》、《生物等效性研究的统计学指导原则》等指导原则。
二、人体生物等效性研究设计
(一)研究类型
采用两制剂、两周期、两序列交叉设计,进行空腹和餐后条件下单次给药的人体生物等效性研究。
(二)受试人群
健康成人受试者。
(三)给药剂量
建议采用申报的最高规格,最大给药剂量不超过0.6g。
(四)给药方法
口服给药。
(五)血样采集
合理设计样品采集时间,使其包含吸收、分布及消除相,同时在给药前采集基线样品。
(六)检测物质
血浆中的乙酰半胱氨酸。
(七)生物等效性评价
以乙酰半胱氨酸的Cmax、AUC0-t和AUC0-∞作为生物等效性评价指标。采用平均生物等效性(Average bioequivalence, ABE)方法进行评价,生物等效性接受标准为受试制剂与参比制剂的Cmax、AUC0-t和AUC0-∞的几何均值比的90%置信区间在80.00%~125.00%范围内。
基于乙酰半胱氨酸基线浓度较低且稳定,如基线浓度不超过Cmax的5%,可不考虑基线带来的影响。
三、人体生物等效性研究豁免
若同时满足以下条件,可豁免低规格制剂的人体生物等效性研究:(1)申报的最高规格制剂符合生物等效性要求;(2)各规格制剂在不同pH介质中体外溶出曲线相似;(3)各规格制剂的处方比例相似。
四、参考文献
1. 国家药品监督管理局.乙酰半胱氨酸颗粒说明书. 2020.
2. 国家药品监督管理局.《以药动学参数为终点评价指标的化学药物仿制药人体生物等效性研究技术指导原则》. 2016.
3. 国家药品监督管理局.《生物等效性研究的统计学指导原则》. 2018.
4. U.S. Food and Drug Administration. Draft Guidance on Acetylcysteine. 2017.
《乙酰半胱氨酸颗粒生物等效性研究技术指导原则(征求意见稿)》起草说明
一、起草目的
乙酰半胱氨酸颗粒是祛痰药类药品,其化学结构中的巯基可使粘蛋白的双硫键断裂,降低痰粘度,使痰容易咳出。临床上用于慢性支气管炎等咳嗽有粘痰而不易咳出的患者。
目前,我国尚无本品生物等效性研究技术指导原则。为进一步规范乙酰半胱氨酸颗粒生物等效性研究,药审中心组织起草了本指导原则,以期为乙酰半胱氨酸颗粒生物等效性研究提供技术指导。
二、起草过程
(一)起草前期调研论证情况
FDA、EMA尚未发布乙酰半胱氨酸颗粒的个药指导原则。FDA于2017年5月发布乙酰半胱氨酸泡腾片(规格2.5g)的BE个药指导原则,研究设计为单剂量、两制剂、交叉体内研究;给药剂量为5g,生物等效性评价对象为乙酰半胱氨酸的90%置信区间。
目前,我国已批准上市的国产乙酰半胱氨酸颗粒(规格0.1g和0.2g)共计7个批准文号,其中包括2个原研地产化批准文号,4个按照新注册分类四类获批上市的仿制药。另有12个新注册分类四类的仿制药上市申请正在审评中。
(二)指导原则制定或修订情况
本指导原则由统计与临床药理学部起草,纳入了中心2023年指导原则制修订计划。根据个药指导原则制定计划,统计与临床药理学部成立工作组,深入调研了美、欧、日等国的生物等效性技术要求及评判标准、审评报告、相关文献等,参考国内既往审评经验,于部门内开展多次讨论,初步形成指导原则初稿。
2022年6月,邀请学术界、产业界等多位一线专家召开改稿会,对关键技术要点进行研讨,并经中心内部征求意见和部门技术委员会审核,形成征求意见稿。
三、起草思路
乙酰半胱氨酸为内源性物质,但体内浓度较低,在生物等效性研究中应关注给药剂量、基线测定、生物等效性评价等。
四、主要内容
本指导原则旨在为乙酰半胱氨酸颗粒生物等效性研究提供技术指导,主要内容包括研究类型、受试人群、给药方法、血样采集、检测物质、生物等效性评价以及豁免等。
本指导原则关键内容为:(1)乙酰半胱氨酸为内源性物质,但体内浓度较低;(2)给药剂量,建议采用申报的最高规格,最大给药剂量不超过0.6g;(3)血样采集,在给药前应采集基线样品;(4)其他方面,基于乙酰半胱氨酸基线浓度较低且稳定,如基线浓度不超过Cmax的5%,可不考虑基线带来的影响。
乙酰半胱氨酸颗粒生物等效性研究应符合本指导原则,还应参照《以药动学参数为终点评价指标的化学药物仿制药人体生物等效性研究技术指导原则》、《生物等效性研究的统计学指导原则》等相关指导原则。
五、需要说明的问题
无。
依托咪酯中/长链脂肪乳注射液生物等效性研究技术指导原则
一、概述
依托咪酯(Etomidate)通过可逆性地阻断11-β-羟化类固醇脱氢酶,从而抑制肾上腺细胞合成皮质醇。依托咪酯中/长链脂肪乳注射液(Etomidate Medium and Long Chain Fat Emulsion Injection)用于成人、6个月以上婴幼儿、儿童和青少年的全身麻醉诱导。
依托咪酯中/长链脂肪乳注射液生物等效性研究应符合本指导原则,还应参照《以药动学参数为终点评价指标的化学药物仿制药人体生物等效性研究技术指导原则》、《生物等效性研究的统计学指导原则》等相关指导原则要求。
二、人体生物等效性研究设计
(一)研究类型
采用两制剂、两周期、两序列交叉设计,进行空腹条件下单次给药的人体生物等效性研究。
(二)受试人群
健康成人受试者。
(三)给药剂量和方法
建议采用5μg/kg/min的给药速率缓慢静脉输注,持续给药30min。
(四)血样采集
合理设计样品采集时间,以充分表征本品药代动力学特征。
(五)检测物质
血浆中的依托咪酯。
(六)生物等效性评价
以依托咪酯的Cmax、AUC0-t和AUC0-∞作为生物等效性评价指标。采用平均生物等效性(Average bioequivalence,ABE)方法进行评价,生物等效性接受标准为受试制剂与参比制剂的Cmax、AUC0-t和AUC0-∞的几何均值比90%置信区间在80.00%~125.00%范围内。
(七)其他
1.输注过程中需由麻醉医生在现场进行监护和必要的干预。
2.研究过程中应监测脑电双频指数(BIS),并提交从给药到给药结束后时间t内BIS值-时间曲线下面积(BISAUC0-t)、给药过程中BIS达到的最低值(BISmin)、最小BIS值出现时间(t-BISmin)等结果。
三、人体生物等效性研究豁免
本品在国内仅上市10mL:20mg规格,本项不适用。
四、参考文献
1. 国家药品监督管理局. 依托咪酯中/长链脂肪乳注射液说明书. 2020.
2. 国家药品监督管理局. 以药动学参数为终点评价指标的化学药物仿制药人体生物等效性研究技术指导原则. 2016.
3. 国家药品监督管理局. 丙泊酚中/长链脂肪乳注射液生物等效性研究技术指导原则. 2021.
4. 国家药品监督管理局. 生物等效性研究的统计学指导原则. 2018.
《依托咪酯中/长链脂肪乳注射液生物等效性研究技术指导原则(征求意见稿)》起草说明
一、背景和目的
依托咪酯(Etomidate)通过可逆性地阻断11-β-羟化类固醇脱氢酶,从而抑制肾上腺细胞合成皮质醇。依托咪酯中/长链脂肪乳注射液(Etomidate Medium and Long Chain Fat Emulsion Injection)用于成人、6个月以上婴幼儿、儿童和青少年的全身麻醉诱导。主要成份为依托咪酯。
目前,我国尚无本品生物等效性研究技术指导原则。为构建以指导原则为核心的审评标准体系,进一步规范依托咪酯中/长链脂肪乳注射液生物等效性研究,药审中心组织起草了本指导原则,以期为依托咪酯中/长链脂肪乳注射液生物等效性研究提供技术指导。
二、起草过程
(一)起草前期调研论证情况
FDA、EMA尚未发布依托咪酯中/长链脂肪乳注射液的个药指导原则。
目前,我国已按照现行《注册管理办法》新注册分类及现行标准批准1个依托咪酯中/长链脂肪乳注射液仿制药上市申请,2个正在审评中,5个已备案开展生物等效性研究。
结合国内已有的研究数据、备案情况等,对依托咪酯中/长链脂肪乳注射液生物等效性研究的设计做出推荐。
(二)指导原则制定或修订情况
本指导原则由统计与临床药理学部起草,纳入了中心2023年指导原则制修订计划。根据个药指导原则制定计划,统计与临床药理学部成立工作组,深入调研了美、欧、日等国的生物等效性技术要求及评判标准、审评报告、相关文献等,参考国内既往审评经验,于部门内开展多次讨论,初步形成指导原则初稿。
2022年6月,邀请学术界、产业界等多位一线专家召开改稿会,对关键技术要点进行研讨,并经中心内部征求意见和部门技术委员会审核,形成征求意见稿。
三、起草思路
依托咪酯中/长链脂肪乳注射液用于成人、6个月以上婴幼儿、儿童和青少年的全身麻醉诱导,属于特殊注射液,在生物等效性研究中应关注给药剂量、方法、生物等效性评价、安全性监测、PD评价等。
四、主要内容
本指导原则旨在为依托咪酯中/长链脂肪乳注射液生物等效性研究提供技术指导,主要内容包括研究类型、受试人群、给药剂量和方法、血样采集、检测物质、生物等效性评价以及其他等。
本指导原则关键内容为:(1)给药剂量和方法部分,建议采用5μg/kg/min的给药速率缓慢静脉输注,持续给药30min;(2)其他方面,建议在输注过程中需由麻醉医生在现场进行监护和必要的干预;(3)其他方面,建议研究过程中应监测脑电双频指数(BIS),并提交BISAUC0-t、BISmin、t-BISmin等结果
依托咪酯中/长链脂肪乳注射液生物等效性研究应符合本指导原则,还应参照《以药动学参数为终点评价指标的化学药物仿制药人体生物等效性研究技术指导原则》、《生物等效性研究的统计学指导原则》等相关指导原则。
五、需要说明的问题
无。
依帕司他片生物等效性研究技术指导原则
一、概述
依帕司他(Epalrestat)是一种可逆性的醛糖还原酶非竞争性抑制剂,对醛糖还原酶具有选择性抑制作用,临床上用于治疗糖尿病性神经病变。根据原研说明书,本品于饭前口服。
依帕司他片生物等效性研究应符合本指导原则要求,还应参照《以药动学参数为终点评价指标的化学药物仿制药人体生物等效性研究技术指导原则》和《生物等效性研究的统计学指导原则》等指导原则。
二、人体生物等效性研究设计
(一) 研究类型
采用两制剂、两周期、两序列交叉设计,进行空腹条件下单次给药的人体生物等效性研究。
(二) 受试人群
健康成人受试者。
(三) 给药剂量
建议采用50mg规格单片服用。
(四) 给药方法
口服给药。
(五) 血样采集
合理设计样品采集时间,使其包含吸收、分布及消除相。
(六) 检测物质
血浆中的依帕司他。
(七) 生物等效性评价
以依帕司他的Cmax、AUC0-t和AUC0-∞作为生物等效性评价指标。采用平均生物等效性(Average bioequivalence, ABE)方法进行评价,生物等效性接受标准为受试制剂与参比制剂的Cmax、AUC0-t和AUC0-∞的几何均值比的90%置信区间在80.00%~125.00%范围内。
三、人体生物等效性研究豁免
本品在国内仅上市50mg规格,此项不适用。
四、 参考文献
1. PMDA. 依帕司他片说明书. 2013.
2. 国家药品监督管理局. 以药动学参数为终点评价指标的化学药物仿制药人体生物等效性研究技术指导原则. 2016.
3. 国家药品监督管理局. 生物等效性研究的统计学指导原则. 2018.
《依帕司他片生物等效性研究技术指导原则(征求意见稿)》起草说明
一、背景和目的
依帕司他(Epalrestat)是一种可逆性的醛糖还原酶非竞争性抑制剂,对醛糖还原酶具有选择性抑制作用,临床上用于治疗糖尿病性神经病变。根据原研说明书,本品于饭前口服。
目前,我国尚无本品生物等效性研究技术指导原则。为构建以指导原则为核心的审评标准体系,进一步规范依帕司他片生物等效性研究,药审中心组织起草了本指导原则,以期为依帕司他片生物等效性研究提供技术指导。
二、起草过程
(一)起草前期调研论证情况
FDA、EMA尚未发布依帕司他片的个药指导原则。
目前,我国已按照现行《注册管理办法》新注册分类及现行标准批准5个依帕司他片仿制药上市申请,6个正在审评中,5个已备案开展生物等效性研究。
结合国内已有的研究数据、备案情况等,对依帕司他片生物等效性研究的设计做出推荐。
(二)指导原则制定或修订情况
本指导原则由统计与临床药理学部起草,纳入了中心2023年指导原则制修订计划。根据个药指导原则制定计划,统计与临床药理学部成立工作组,深入调研了美、欧、日等国的生物等效性技术要求及评判标准、审评报告、相关文献等,参考国内既往审评经验,于部门内开展多次讨论,初步形成指导原则初稿。
2022年6月,邀请学术界、产业界等多位一线专家召开改稿会,对关键技术要点进行研讨,并经中心内部征求意见和部门技术委员会审核,形成征求意见稿。
三、起草思路
依帕司他片用于治疗糖尿病性神经病变,根据原研说明书,本品于饭前口服,在生物等效性研究中应关注研究类型等。
四、主要内容
本指导原则旨在为依帕司他片生物等效性研究提供技术指导,主要内容包括研究类型、受试人群、给药剂量和方法、血样采集、检测物质、生物等效性评价以及其他等。
本指导原则关键内容为:研究类型部分建议采用两制剂、两周期、两序列交叉设计,进行空腹条件下单次给药的人体生物等效性研究。
依帕司他片生物等效性研究应符合本指导原则,还应参照《以药动学参数为终点评价指标的化学药物仿制药人体生物等效性研究技术指导原则》、《生物等效性研究的统计学指导原则》等相关指导原则。
五、需要说明的问题
无。
硫辛酸片生物等效性研究技术指导原则
一、概述
硫辛酸(Thioctic Acid)为B族维生素,是丙酮酸脱氢酶系和α-酮戊二酸脱氢酶系的辅酶,临床上用于治疗糖尿病多发性周围神经病变。根据原研说明书,本品需于早餐前半小时服用。
硫辛酸片生物等效性研究应符合本指导原则要求,还应参照《以药动学参数为终点评价指标的化学药物仿制药人体生物等效性研究技术指导原则》、《生物等效性研究的统计学指导原则》、《高变异药物生物等效性研究技术指导原则》等指导原则。
二、人体生物等效性研究设计
(一) 研究类型
采用两制剂、两周期、两序列交叉设计,也可采用部分重复或完全重复交叉设计,进行空腹条件下单次给药的人体生物等效性研究。
(二) 受试人群
健康成人受试者。
(三) 给药剂量
建议采用申报的最高规格单片服用。
(四) 给药方法
口服给药。
(五) 血样采集
合理设计样品采集时间,使其包含吸收、分布及消除相。
(六) 检测物质
血浆中的硫辛酸。
(七) 生物等效性评价
以硫辛酸的Cmax、AUC0-t和AUC0-∞作为生物等效性评价指标。如采用平均生物等效性(Average bioequivalence, ABE)方法进行评价,受试制剂与参比制剂的Cmax、AUC0-t和AUC0-∞的几何均值比的90%置信区间在80.00%~125.00%范围内。如采用参比制剂标度的平均生物等效性(Reference-scaled average bioequivalence, RSABE)方法进行评价,参比制剂的个体内标准差(SWR)应大于等于0.294,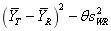 的单侧95%置信区间上限小于等于零,同时,制剂间主要药动学参数的几何均值比(Geometric mean ratio, GMR)的点估计值应在80.00%~125.00%范围内。
的单侧95%置信区间上限小于等于零,同时,制剂间主要药动学参数的几何均值比(Geometric mean ratio, GMR)的点估计值应在80.00%~125.00%范围内。
三、人体生物等效性研究豁免
若同时满足以下条件,可豁免低规格制剂的人体生物等效性研究:(1)申报的最高规格制剂符合生物等效性要求;(2)各规格制剂在不同pH介质中体外溶出曲线相似;(3)各规格制剂的处方比例相似。
四、参考文献
1. 国家药品监督管理局.以药动学参数为终点评价指标的化学药物仿制药人体生物等效性研究技术指导原则. 2016.
2. 国家药品监督管理局.生物等效性研究统计学指导原则.2018.
3. 国家药品监督管理局.高变异药物生物等效性研究技术指导原则.2018.
《硫辛酸片生物等效性研究技术指导原则(征求意见稿)》起草说明
一、背景和目的
硫辛酸(Thioctic Acid)为B族维生素,是丙酮酸脱氢酶系和α-酮戊二酸脱氢酶系的辅酶,临床上用于治疗糖尿病多发性周围神经病变。
目前,我国尚无本品生物等效性研究技术指导原则。为构建以指导原则为核心的审评标准体系,进一步规范硫辛酸片生物等效性研究,药审中心组织起草了本指导原则,以期为硫辛酸片生物等效性研究提供技术指导。
二、起草过程
(一)起草前期调研论证情况
FDA、EMA尚未发布硫辛酸片的个药指导原则。
目前,我国尚无原研厂家的产品上市,国内有2家仿制药企业获批。另有1个新注册分类3类的仿制药上市申请正在审评中。
结合国内已有的研究数据、备案情况等,对硫辛酸片生物等效性研究的设计做出推荐。
(二)指导原则制定或修订情况
本指导原则由统计与临床药理学部起草,纳入了中心2023年指导原则制修订计划。根据个药指导原则制定计划,统计与临床药理学部成立工作组,深入调研了美、欧、日等国的生物等效性技术要求及评判标准、审评报告、相关文献等,参考国内既往审评经验,于部门内开展多次讨论,初步形成指导原则初稿。
2022年6月,邀请学术界、产业界等多位一线专家召开改稿会,对关键技术要点进行研讨,并经中心内部征求意见和部门技术委员会审核,形成征求意见稿。
三、起草思路
硫辛酸片需于早餐前半小时服用,且存在高变异的情况,在生物等效性研究中应关注研究类型、生物等效性评价方法等。
四、主要内容
本指导原则旨在为硫辛酸片生物等效性研究提供技术指导,主要内容包括研究类型、受试人群、给药剂量和方法、血样采集、检测物质、生物等效性评价以及豁免等。
本指导原则关键内容为:(1)研究类型部分,建议采用两制剂、两周期、两序列交叉设计,也可采用部分重复或完全重复交叉设计,进行空腹条件下单次给药的人体生物等效性研究;(2)给药剂量,建议采用申报的最高规格;(3)生物等效性评价方面,以硫辛酸的Cmax、AUC0-t和AUC0-∞作为生物等效性评价指标。如采用平均生物等效性方法进行评价,受试制剂与参比制剂的Cmax、AUC0-t和AUC0-∞的几何均值比的90%置信区间在80.00%~125.00%范围内。如采用参比制剂标度的平均生物等效性方法进行评价,参比制剂的个体内标准差(SWR)应大于等于0.294,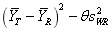 的单侧95%置信区间上限小于等于零,同时,制剂间主要药动学参数的几何均值比的点估计值应在80.00%~125.00%范围内。
的单侧95%置信区间上限小于等于零,同时,制剂间主要药动学参数的几何均值比的点估计值应在80.00%~125.00%范围内。
硫辛酸片生物等效性研究应符合本指导原则,还应参照《以药动学参数为终点评价指标的化学药物仿制药人体生物等效性研究技术指导原则》、《生物等效性研究的统计学指导原则》和《高变异药物生物等效性研究技术指导原则》等相关指导原则。
五、需要说明的问题
无。
利丙双卡因乳膏生物等效性研究技术指导原则
一、概述
利丙双卡因乳膏(Lidocaine and Prilocaine Cream)是利多卡因和丙胺卡因按重量比1:1混合制备而成的复方乳膏剂,用于针穿刺、浅层外科手术的皮肤局部麻醉。
利丙双卡因乳膏人体生物等效性研究应符合本指导原则要求,还应参照《皮肤外用化学仿制药研究技术指导原则(试行)》、《以药动学参数为终点评价指标的化学药物仿制药人体生物等效性研究技术指导原则》、《生物等效性研究的统计学指导原则》等指导原则。
二、人体生物等效性研究设计
(一)研究类型
采用两制剂、两周期、两序列交叉设计,进行空腹条件下的单次皮肤给药的人体生物等效性研究。
(二)受试人群
健康成人受试者。
(三)给药剂量
根据原研进口产品说明书,成人每10平方厘米约给药1.5g。建议单次给药总量不超过60g。
(四)给药方法
皮肤局部涂抹。
(五)血样采集
合理设计样品采集时间,使其包含吸收、分布及消除相。
(六)检测物质
血浆中的利多卡因、丙胺卡因。
(七)生物等效性评价
以利多卡因、丙胺卡因的Cmax、AUC0-t、AUC0-∞作为生物等效性研究评价指标。采用平均生物等效性(Average bioequivalence, ABE)方法进行评价,生物等效性接受标准为受试制剂与参比制剂的Cmax、AUC0-t和AUC0-∞的几何均值比90%置信区间在80.00%~125.00%范围内。
(八)其他
1、若Q1、Q2不一致(但关键辅料一致),Q3一致的前提下,可以采用PK-BE研究桥接与参比制剂的一致性进行上市申请。
2、受试者给药前给药部位皮肤应完好无损。
3、应在本研究中同时观察受试制剂与参比制剂的皮肤反应性,并评价两制剂皮肤反应的可比性。观察局部皮肤反应应包括以下几点:苍白或发白、红斑、温感、水肿和皮疹等。应记录反应程度和缓解时间,并将观察结果制成表格。
三、人体生物等效性研究豁免
本项不适用。
四、参考文献
1. 国家药品监督管理局. 利丙双卡因乳膏(恩纳)说明书. 2019.
2. 国家药品监督管理局. 以药动学参数为终点评价指标的化学药物仿制药人体生物等效性研究技术指导原则. 2016.
3. 国家药品监督管理局. 生物等效性研究的统计学指导原则. 2018.
4. 国家药品监督管理局. 皮肤外用化学仿制药研究技术指导原则(试行). 2021.
5. U.S. Food and Drug Administration. Draft Guidance on Lidocaine; Prilocaine. 2014.
《利丙双卡因乳膏生物等效性研究技术指导原则(征求意见稿)》起草说明
一、背景和目的
利丙双卡因乳膏(Lidocaine and Prilocaine Cream)是利多卡因和丙胺卡因按重量比1:1混合制备而成的复方乳膏剂,用于针穿刺、浅层外科手术的皮肤局部麻醉。
目前,我国尚无本品生物等效性研究技术指导原则。为构建以指导原则为核心的审评标准体系,进一步规范利丙双卡因乳膏生物等效性研究,药审中心组织起草了本指导原则,以期为利丙双卡因乳膏生物等效性研究提供技术指导。
二、起草过程
(一)起草前期调研论证情况
FDA已发布利丙双卡因乳膏生物等效性研究的个药指导原则。
目前,我国已按照现行《注册管理办法》新注册分类及现行标准批准2个利丙双卡因乳膏仿制药上市申请,1个利丙双卡因乳膏补充申请,18个正在审评中,17个已备案开展生物等效性研究。
结合国内已有的研究数据、备案情况等,对利丙双卡因乳膏生物等效性研究的设计做出推荐。
(二)指导原则制定或修订情况
本指导原则由统计与临床药理学部起草,纳入了中心2023年指导原则制修订计划。根据个药指导原则制定计划,统计与临床药理学部成立工作组,深入调研了美、欧、日等国的生物等效性技术要求及评判标准、审评报告、相关文献等,参考国内既往审评经验,于部门内开展多次讨论,初步形成指导原则初稿。
2022年8月,邀请学术界、产业界等多位一线专家召开改稿会,对关键技术要点进行研讨,并经中心内部征求意见和部门技术委员会审核,形成征求意见稿。
三、起草思路
利丙双卡因乳膏为皮肤制剂,在生物等效性研究中应关注给药剂量、给药方法、血样采集及生物等效性评价等。
四、主要内容
本指导原则旨在为利丙双卡因乳膏生物等效性研究提供技术指导,主要内容包括研究类型、受试人群、给药剂量、给药方法、血样采集、检测物质、生物等效性评价以及其他等。
本指导原则关键内容为:(1)研究类型部分,建议采用两制剂、两周期、两序列交叉设计,进行空腹条件下的单次皮肤给药的人体生物等效性研究;(2)给药剂量,建议根据原研进口产品说明书,成人每10平方厘米约给药1.5g。建议单次给药总量不超过60g;(3)给药方法,建议皮肤局部涂抹。
利丙双卡因乳膏生物等效性研究应符合本指导原则,还应参照《皮肤外用化学仿制药研究技术指导原则(试行)》、《以药动学参数为终点评价指标的化学药物仿制药人体生物等效性研究技术指导原则》、《生物等效性研究的统计学指导原则》等相关指导原则。
五、需要说明的问题
无。
相关推荐
CIO提供以下相关文库下载、合规服务以及线上培训课程学习。

E邀专家

















粤公网安备 44010402001640号 | 互联网药品信息服务资格证编号:(粤)-经营性-2018-0045 | 增值电信许可证:粤B2-20261803 | 粤ICP备11100335号
违法和不良信息举报邮箱:info@ciopharma.com | ©2003-2026 ciopharma.com 广东国健医药咨询有限公司 版权所有

